Bardet-Biedl-Syndrom · Center of Excellence
Das Bardet-Biedl-Syndrom (BBS)
Interdisziplinäre Diagnostik, Forschung und Versorgung im Center of Excellence
Das Bardet-Biedl-Syndrom (BBS) ist eine seltene, genetisch bedingte Multisystemerkrankung aus der Gruppe der Ziliopathien, die durch eine Fehlfunktion der Zilien, winziger Zellfortsätze, verursacht wird. Sie betrifft zahlreiche Organsysteme gleichzeitig und erfordert daher eine hochspezialisierte, interdisziplinäre Betreuung.
Unser Center of Excellence vereint klinische Expertise, moderne Diagnostik und international vernetzte Forschung – mit dem Ziel, die Versorgung von Menschen mit BBS und ähnlichen Ziliopathien nachhaltig zu verbessern.
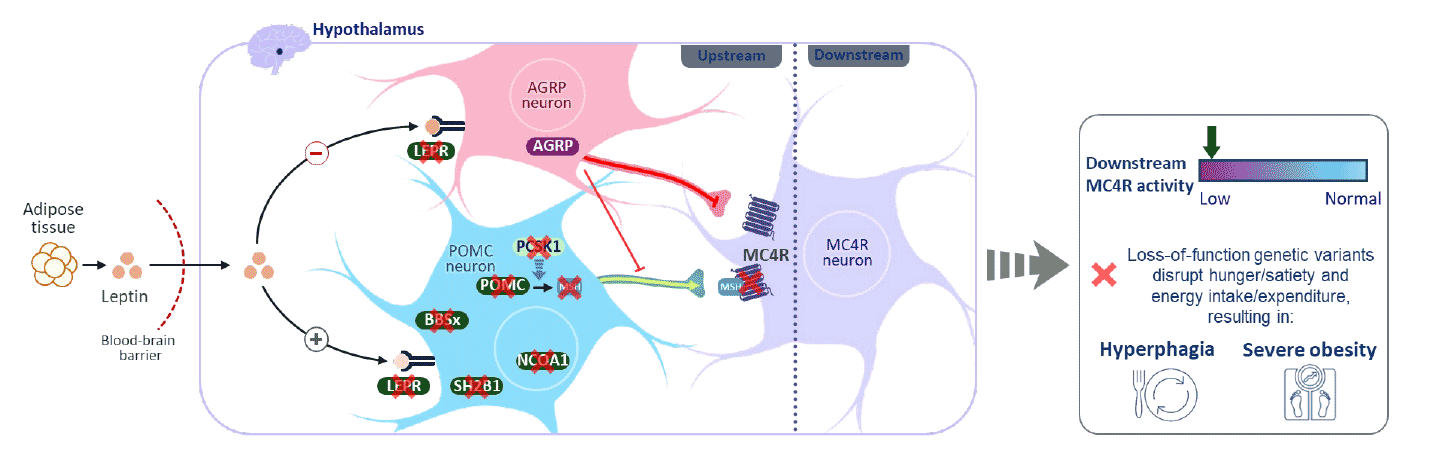
BBS entsteht durch genetische Veränderungen, die die Funktion der Zilien stören – mikroskopisch kleiner Zellfortsätze, die für Signalprozesse im Körper entscheidend sind. Besonders betroffen sind die Sehfunktion, Stoffwechselregulation, Nierenfunktion und hormonelle Entwicklung. Kardinalsymptome eines BBS sind die Netzhautdystrophie (Retinitis pigmentosa), eine Hyperphagie-assoziierte Adipositas, Nierenauffälligkeiten, Polydaktylie (überschüssige Finger und Zehen), Fehlentwicklung der Geschlechtsorgane und -hormone sowie Verhaltens- und Entwicklungsstörungen. Die Erkrankung zeigt jedoch eine hohe klinische Variabilität, was die Diagnose häufig verzögert.
BBS erfordert das eng abgestimmte Zusammenspiel von spezialisierter Versorgung und Spitzenforschung. PatientInnen mit BBS sehen sich mit charakteristischen Merkmalen wie übermäßigem Essen sowie daraus resultierender Fettleibigkeit, Netzhautdegeneration und Niereninsuffizienz konfrontiert. Die Behandlung von BBS benötigt daher eine koordinierte Strategie, die über die reine Symptombehandlung deutlich hinausgeht.

Das BBS Center of Excellence versammelt daher ein interdisziplinäres Team, das sich der Erforschung von Zusammenhängen zwischen Endokrinologie, Genetik, Neurowissenschaften und Stoffwechsel, mit dem Folkus auf deren Regulation über die Melanocortin-Rezeptor-Signalwege widmet. Auch in der umfassenden Versorgung von Patientinnen und Patienten mit BBS sowie verwandten seltenen genetischen Erkrankungen arbeiten Ärztinnen und Ärzte ver-schiedener Fachrichtungen gemeinsam mit spezialisierten Pflegekräften, Therapeutinnen, Wissenschaftlern und Koordinatoren zusammen. Forschung, Diagnostik und Therapie werden durch die enge Kooperation mit internationalen Expertengruppen ständig verbessert. Das BBS Center of Excellence begleitet Betroffene sowie ihre Familien langfristig und individuell. In die Versorgung fließen die neuesten wissenschaftlichen Erkenntnisse mit ein.
Fakten zum Bardet-Biedl-Syndrom
- Diagnose: Die Diagnose BBS gestaltet sich oft schwierig und erfolgt spät aufgrund der variablen Symptome. Eine gesicherte Diagnose wird durch molekulargenetische Untersuchungen, insbesondere Next Generation Sequencing, gestellt. Pathogene Varianten in den BBS-Genen 1, 7 und 10 sind weltweit am häufigsten.
- Vererbung:* BBS wird autosomal-rezessiv vererbt. Als krankheitsauslösend sind aktuell über 26 unterschiedliche Mutationen auf unterschiedlichen Genen bekannt. Die Zahl der detektierten Gene steigt zuletzt immer weiter. Viele dieser Gene kodieren Proteine, die zusammen den BBSome bilden oder dessen Funktion unterstützen.
- Häufigkeit: BBS ist eine seltene Erkrankung mit einer Prävalenz von etwa 1:125.000 bis 1:175.000 in Europa. In isolierten Populationen wie z. B. Kuwait oder Neufundland (1:18.000) ist sie deutlich höher. Durch verzögerte Diagnosestellung sowie klinischer und genetischer Variabilität ist die Dunkelziffer vermutlich hoch. Inzidenz erhöht das Risiko für die Entwicklung von autosomal-rezessiven Erkrankungen.
Symptome
Nierenbeteiligung
> 50%
Hyperphagie und Adipositas
> 80%
Hypogonadismus
> 50% (weiblich seltener)
Netzhautdegeneration
> 90%
Entwicklungsverzögerung
> 50%
Polydaktylie
> 60%
Sonstige
(unter anderem Anosmie, Leberfibrose, Fettleber, Herzbeteiligung, Hörstörungen, Zahnauffälligkeiten, Morbus Hirschsprung)
0
Zeitpunkt des Auftretens (Lebensalter in Jahren)
Mittlerer Zeitpunkt der Diagnosestellung des BBS: 5,6 ± 4,8 LJ (8–19)
18
Cetiner, M., Pape, L., König, J. et al. Das Bardet-Biedl-Syndrom. Monatsschr Kinderheilkd (2024).
https://doi.org/10.1007/s00112-024-02030-7