Bardet-Biedl-Syndrome · Center of Excellence
The Bardet-Biedl-Syndrome (BBS)
Interdisciplinary diagnostics, research and care at the Centre of Excellence
Bardet-Biedl syndrome (BBS) is a rare, genetically caused multisystem disorder belonging to the group of ciliopathies, which is caused by a malfunction of the cilia, tiny cellular projections. It affects numerous organ systems simultaneously and therefore requires highly specialised, interdisciplinary care.
Our Centre of Excellence combines clinical expertise, modern diagnostics and internationally networked research – with the aim of sustainably improving care for people with BBS and similar ciliopathies.
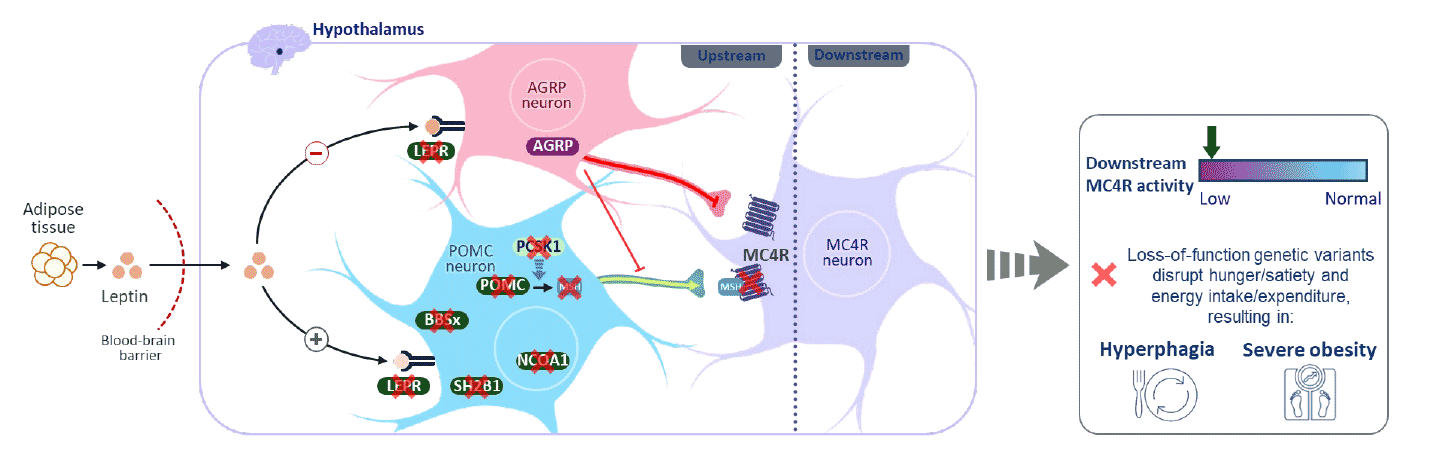
BBS is caused by genetic changes that disrupt the function of cilia – microscopic cell projections that are crucial for signalling processes in the body. Vision, metabolic regulation, kidney function and hormonal development are particularly affected. The cardinal symptoms of BBS are retinal dystrophy (retinitis pigmentosa), hyperphagia-associated obesity, renal abnormalities, polydactyly (extra fingers and toes), malformation of the reproductive organs and hormones, as well as behavioural and developmental disorders. However, the condition exhibits a high degree of clinical variability, which often delays diagnosis.
BBS requires the close coordination of specialised care and cutting-edge research. Patients with BBS face characteristic symptoms such as overeating and the resulting obesity, retinal degeneration and renal insufficiency. The treatment of BBS therefore requires a coordinated strategy that goes well beyond mere symptom management.

The BBS Centre of Excellence therefore brings together an interdisciplinary team dedicated to researching the links between endocrinology, genetics, neuroscience and metabolism, with a focus on their regulation via the melanocortin receptor signalling pathways. Doctors from various disciplines also work together with specialist nurses, therapists, scientists and coordinators to provide comprehensive care for patients with BBS and related rare genetic disorders. Research, diagnostics and therapy are constantly being improved through close cooperation with international expert groups. The BBS Centre of Excellence provides long-term, personalised support to those affected and their families. The latest scientific findings are incorporated into patient care.
Facts about Bardet-Biedl syndrome
- Diagnosis: Diagnosing BBS is often difficult and is made late due to the variable symptoms. A definitive diagnosis is established through molecular genetic testing, in particular next-generation sequencing. Pathogenic variants in the BBS genes 1, 7 and 10 are the most common worldwide.
- Inheritance:* BBS is inherited in an autosomal recessive manner. Over 26 different mutations in various genes are currently known to cause the condition. The number of identified genes has been steadily increasing recently. Many of these genes encode proteins that together form the BBSome or support its function.
- Prevalence: BBS is a rare disorder with a prevalence of approximately 1 in 125,000 to 1 in 175,000 in Europe. In isolated populations such as Kuwait or Newfoundland (1:18,000), it is significantly higher. Due to delayed diagnosis and clinical and genetic variability, the number of unreported cases is likely to be high. The incidence increases the risk of developing autosomal recessive disorders.
Symptoms
Kidney involvement
> 50%
Hyperphagia and obesity
> 80%
Hypogonadism
> 50% (less common in females)
Retinal degeneration
> 90%
Developmental delay
> 50%
Polydactyly
> 60%
Other
(including anosmia, liver fibrosis, fatty liver, cardiac involvement, hearing impairments, dental abnormalities, Hirschsprung’s disease)
0
Age at onset (in years)
Average age at diagnosis of BBS: 5,6 ± 4,8 years (8–19)
18
Cetiner, M., Pape, L., König, J. et al. Das Bardet-Biedl-Syndrom. Monatsschrift Kinderheilkunde (2024).
https://doi.org/10.1007/s00112-024-02030-7